
la Storia
Il primo paziente, affetto da una sindrome con caratteristiche analoghe a quelle presenti nella Sindrome di Williams (SW), è stato descritto nel 1952 dal pediatra italo-svizzero Guido Fanconi.
Il paziente presentava un aumento dei livelli ematici del calcio (ipercalcemia idiopatica) e un restringimento dell'aorta (stenosi sopravalvolare dell'aorta o SSVA). Questa prima osservazione non ha avuto seguito fino agli anni '60, quando due pediatri, il neozelandese Williams e l'americano Beuren, hanno delineato la sindrome e ne hanno descritto le principali caratteristiche. Per questo, la sindrome viene oggi identificata con il cognome di questi due autori, o più semplicemente, con il primo di loro.
William e Beuren hanno definito le caratteristiche facciali della sindrome, che di solito sono bene evidenti già nei primi mesi di vita, e si associano spesso alla SSVA o ad un'altra cardiopatia. Le figure A e B riproducono lo stesso bambino con SW, all'età di 1 e 4 anni. Nella vecchia letteratura, le caratteristiche del viso di queste persone erano state paragonate a quelle delle figure mitologiche dei fauni (elfin face).

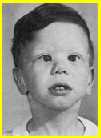
La figura C riproduce un ragazzo di 20 anni, che documenta l'evoluzione nel tempo di alcune caratteristiche cliniche della sindrome.

Per molti anni sono rimaste ignote le basi biologiche e molecolari della SW. Di fatto, la maggior parte di questi casi si presenta in maniera isolata (sporadica) nella famiglia e, in queste condizioni, risulta, per definizione, più difficile l'analisi genetica.
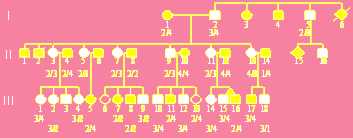
La definizione dell'origine della SW ha tratto vantaggio dalla sua associazione con la SSVA. La SSVA può essere ereditata nelle famiglie come difetto anatomico isolato, ad "eredit autosomica dominante".
In pratica, la malattia si trasmette da un genitore affetto in media al 50% dei figli, indipendentemente dal loro sesso, così come nell'albero genealogico riprodotto nella Figura successiva:
Dal punto di vista anatomo-patologico, la SSVA consiste in un restringimento dell'aorta, immediatamente al di sotto della valvola omonima, a livello della regione di fuoriuscita dell'aorta dal cuore (Figure).

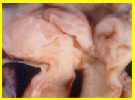
Stenosi sopravalvolare dell'aorta
1993 la svolta
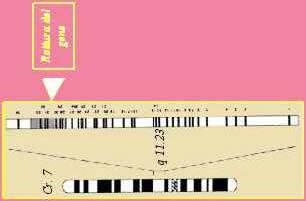
L'osservazione critica nel definire il meccanismo molecolare della SSVA avvenuta nel 1993, quando stata studiata una famiglia di quattro generazioni, nella quale tutti i pazienti con la cardiopatia erano anche portatori di un'anomalia cromosomica, consistente nella traslocazione bilanciata e reciproca (trasferimento di parti di cromosoma tra cromosomi diversi), tra il braccio lungo del cromosoma 7 e il braccio corto del cromosoma 6.
Il punto di rottura sul cromosoma 7 è stato localizzato nella banda 7q11.23. L'associazione tra la traslocazione e la SSVA in tutti i soggetti di questa famiglia, in accordo con le precedenti evidenze che avevano localizzato il gene-malattia sul cromosoma 7, ha suggerito un nesso di causalita' tra questi due eventi ed ha accelerato le ricerche rivolte all'identificazione del difetto molecolare.
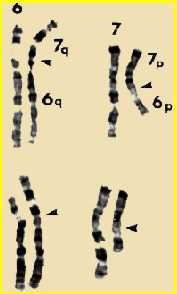
Utilizzando tecniche di biologia molecolare, stato possibile eseguire un'analisi fine della regione 7q11.23, interrotta dalla traslocazione, e dimostrare che aveva "rotto" il gene dell'elastina (ELN).
Questo studio ha suggerito che la mutazione del gene ELN fosse correlata con la SSVA nella famiglia.
L'osservazione è stata confermata su altri pazienti con SSVA isolata, non associata ad anomalie cromosomiche visibili. Un ulteriore aspetto, emerso dall'analisi della famiglia che presentava la traslocazione 6;7 associata a SSVA, è stato il rilievo di segni clinici sfumati della SW nel paziente pi giovane della IV generazione, IV.T2.
Questo dato ha indicato che anche la SW poteva essere correlata ad una mutazione del gene ELN.
Nel 1993 è stata avviata l'analisi sistematica di ELN su un gruppo di pazienti con SW sporadica e in alcuni casi familiari, nei quali la malattia era trasmessa da un genitore affetto.
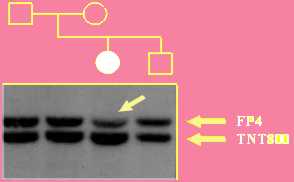
In tutti i casi è stato dimostrato che uno dei due cromosomi 7 era deleto a livello del gene ELN, ci mancava uno dei due geni.
La mutazione dei pazienti con SW veniva allora identificata per "effetto di dose".
In pratica, il paziente presenta, rispetto ai suoi familiari non affetti, una riduzione di circa il 50% del segnale del gene ELN (indicato come FP4), rispetto ad una sonda di controllo (TNT800).
Questi risultati hanno dimostrato un rapporto causale tra la delezione di ELN e la SW.
Nuovi metodi di analisi
Negli anni successivi, l'analisi della delezione è stata semplificata.
La conferma del sospetto diagnostico della SW viene fornita dall'ibridazione in situ fluorescente (Fluorescent In Situ Hybridization - FISH), utilizzando una sonda specifica della regione deleta.
Le analisi molecolari hanno permesso, negli ultimi anni, di analizzare dettagliatamente la regione 7q11.23 interessata dalla delezione in oltre il 95% delle persone con SW.
Questi studi hanno dimostrato che la delezione che caratterizza i pazienti, oltre ad eliminare il gene ELN, li rende emizigoti (deleti) per numerosi altri geni della regione. Attualmente sono stati identificati in questa area genomica, che si estende per circa un milione e mezzo di basi (1,5 Mb), una cinquantina di geni.